
Bioequivalence Studies: What the FDA Requires Generic Drug Manufacturers to Prove
When you pick up a generic pill at the pharmacy, you expect it to work just like the brand-name version. But how does the FDA make sure it does? The answer lies in bioequivalence studies - the scientific backbone of generic drug approval in the U.S.
Why Bioequivalence Matters
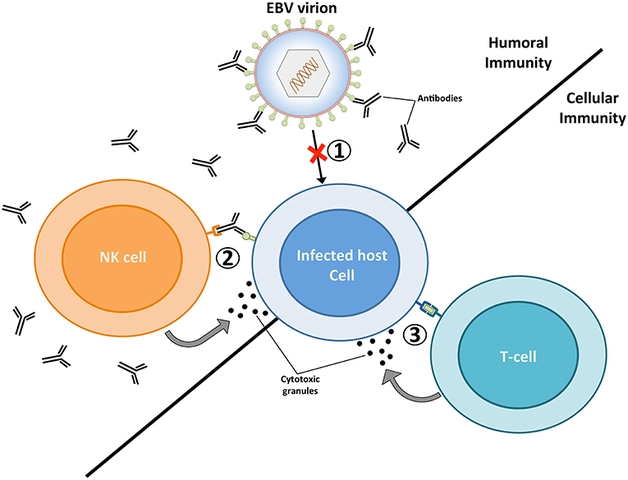
Generic drugs save patients and the healthcare system billions each year. In 2023, they made up 90% of all prescriptions filled in the U.S., yet cost only 23% of what brand-name drugs do. But cost savings shouldn’t mean risk. The FDA doesn’t just accept a generic drug because it has the same active ingredient. It demands proof that the drug behaves the same way in the body.This proof comes from bioequivalence studies. These aren’t just lab tests or paperwork. They’re clinical trials conducted on real people - healthy volunteers - to measure exactly how the body absorbs and processes the drug. If the generic drug delivers the same amount of medicine at the same speed as the brand-name version, it’s considered bioequivalent. And that means it’s therapeutically equivalent.
The Core Requirements: AUC and Cmax
The FDA doesn’t guess whether a drug works the same. It measures it. Every bioequivalence study must track two key numbers: area under the concentration-time curve (AUC) and maximum plasma concentration (Cmax).AUC tells you how much of the drug enters the bloodstream over time - the total exposure. Cmax tells you how high the drug’s concentration peaks in the blood. These numbers are calculated from blood samples taken at regular intervals after a single dose.
The FDA’s rule is strict: the 90% confidence interval for the ratio of the generic to the brand-name drug’s AUC and Cmax must fall between 80% and 125%. This is known as the 80/125 rule. It’s been in place since 1992 and hasn’t changed. Why? Because decades of data show that if two drugs meet this standard, patients will get the same clinical results.
For example, if the brand-name drug reaches a Cmax of 100 ng/mL, the generic must reach between 80 and 125 ng/mL. The same applies to AUC. If it falls outside that range, the FDA will reject the application - no exceptions.
Who Gets Tested and Under What Conditions?
Bioequivalence studies typically involve 24 to 36 healthy adult volunteers. They’re not patients with the disease - they’re healthy to eliminate confounding variables. The study is usually done in two phases: first, participants take the generic drug after fasting; then, after a washout period, they take the brand-name drug under the same conditions.But fasting isn’t always enough. Some drugs are affected by food. If the brand-name drug’s label says “take with food,” the FDA requires a second study under fed conditions. For drugs like ketoconazole or atazanavir, food can change absorption dramatically. If the generic behaves differently with food than the brand, it won’t be approved.
All studies must follow Good Laboratory Practice (GLP) rules. That means every blood sample is tracked, stored correctly, and analyzed with validated methods. The FDA checks this closely. A single mistake in sample handling can invalidate an entire study.

When Can You Skip the Human Study? Biowaivers
Not every generic drug needs a full clinical trial. The FDA allows biowaivers - exceptions where manufacturers can skip human testing. These apply to a limited set of products where absorption is predictable and not affected by formulation changes.Biowaivers are granted under the Q1-Q2-Q3 framework:
- Q1: Same active ingredient and identical inactive ingredients (excipients) as the brand-name drug.
- Q2: Same dosage form, strength, and route of administration.
- Q3: Same pH, solubility, and dissolution profile as the reference drug.
Examples include oral solutions like amoxicillin, eye drops like ciprofloxacin, and certain inhaled anesthetics. Topical products meant to act locally - like hydrocortisone cream - can use in vitro tests instead of blood draws. The FDA accepts in vitro release testing (IVRT) and in vitro permeation testing (IVPT) if they prove the drug releases the same way from the cream as the brand.
Biowaivers can cut development time by 6 to 12 months and save companies hundreds of thousands of dollars. But they’re not easy to get. The FDA has approved biowaivers for only about 1,200 product types as of 2023 - and each one has its own detailed requirements.
Tighter Rules for High-Risk Drugs
Not all drugs are created equal. Some have a narrow therapeutic index (NTID) - meaning the difference between a safe dose and a toxic one is tiny. For drugs like warfarin, levothyroxine, or phenytoin, even a small change in absorption can cause serious harm.For these, the FDA doesn’t use the standard 80/125 rule. Instead, it requires a tighter range: 90% to 111% for both AUC and Cmax. This reduces the chance of under- or overdosing.
These drugs also require more complex study designs. Sometimes, multiple doses are tested. Sometimes, patients with the condition are included instead of healthy volunteers. The FDA reviews these applications with extra scrutiny - and rejections are more common.
What Happens When Studies Fail?
Despite the clear rules, many generic applications get rejected on first review. In 2022, only 43% of ANDA submissions were approved on the first try. The most common reasons? Poor study design, too few participants, unreliable lab methods, or incomplete documentation.Companies that follow the FDA’s Product-Specific Guidance (PSG) documents - which detail exact testing requirements for each drug - have a 68% first-cycle approval rate. Those that don’t? Just 29%.
Each PSG is tailored. For example, the PSG for metformin ER tablets requires a specific dissolution profile and fasting/fed study design. For topical tacrolimus ointment, the FDA requires in vitro permeation data plus a clinical endpoint study. Ignoring these documents is like building a house without blueprints.

The Future: New Tools and Complex Drugs
The FDA is updating its approach for newer, harder-to-copy drugs. Topical creams, inhalers, and drug-device combos (like insulin pens) are rising in complexity. Traditional bioequivalence methods don’t always work.That’s why the FDA is turning to advanced tools:
- Physiologically Based Pharmacokinetic (PBPK) modeling: Computer simulations that predict how a drug behaves based on its chemistry and body physiology.
- Quantitative systemic exposure comparisons: For topical drugs, measuring actual drug levels in the skin instead of blood.
- Advanced in vitro models: Artificial skin or gut membranes that mimic human absorption.
The FDA plans to release draft guidances for 45 complex drug categories by mid-2024. These will help manufacturers design better studies and avoid costly delays.
Why This System Works
The U.S. generic drug system is one of the most successful in the world. It balances safety, science, and affordability. The bioequivalence requirement isn’t perfect - critics say the 80/125 rule is too broad for some drugs - but it’s backed by decades of real-world evidence.Patients get the same medicine at a fraction of the cost. Manufacturers have clear rules to follow. And the FDA has tools to catch problems before drugs hit the market.
It’s not about cutting corners. It’s about proving, with science, that a cheaper pill does exactly what it’s supposed to do.
What is bioequivalence in the context of generic drugs?
Bioequivalence means that a generic drug delivers the same amount of active ingredient into the bloodstream at the same rate as the brand-name drug. This ensures the generic works the same way in the body, producing identical therapeutic effects and safety outcomes when used under the same conditions.
What are the FDA’s acceptance criteria for bioequivalence?
The FDA requires that the 90% confidence interval for the ratio of the generic to brand-name drug’s AUC and Cmax falls between 80% and 125%. For narrow therapeutic index drugs like warfarin or levothyroxine, the range is tighter: 90% to 111%.
Do all generic drugs need human bioequivalence studies?
No. Some products qualify for biowaivers based on the Q1-Q2-Q3 criteria: identical active and inactive ingredients, same dosage form, and matching physicochemical properties. Examples include oral solutions, eye drops, and certain topical creams. In vitro tests can replace human studies for these.
Why are bioequivalence studies done in healthy volunteers?
Healthy volunteers eliminate disease-related variables that could affect drug absorption. This gives a clearer picture of how the formulation itself influences bioavailability. For drugs with narrow therapeutic windows, studies may include patients, but this is the exception.
What happens if a generic drug fails bioequivalence testing?
The FDA issues a complete response letter outlining deficiencies. The manufacturer must fix the study design, conduct new trials, and resubmit. This can delay approval by months or even years. About 57% of ANDA applications are rejected on first submission, often due to poor study design or failure to follow product-specific guidance.
How long do bioequivalence studies take to complete?
A typical study lasts 4 to 8 weeks, including screening, dosing, and follow-up. The actual clinical phase - where volunteers take the drug and give blood samples - usually takes 1 to 2 weeks per treatment period. But preparing the study, analyzing data, and writing the report can add several months.
Are bioequivalence standards the same in the U.S. and Europe?
Yes. The FDA and the European Medicines Agency (EMA) have aligned their bioequivalence requirements under the International Council for Harmonisation (ICH). As of 2023, 87% of their standards are identical, including the 80/125 rule and use of AUC and Cmax as primary endpoints.
Can a generic drug be approved without being bioequivalent to the brand-name version?
No. The FDA requires both pharmaceutical equivalence (same active ingredient, strength, dosage form) and bioequivalence. Without bioequivalence, the drug cannot be approved as a generic under the Abbreviated New Drug Application (ANDA) process. There are no exceptions.
Next Steps for Manufacturers
If you’re developing a generic drug, start by finding the FDA’s Product-Specific Guidance for your drug. There are over 2,100 available. Follow them exactly. Don’t assume your formulation is “close enough.” The FDA doesn’t work on assumptions - it works on data.Invest in reliable analytical methods. Use validated assays. Recruit enough volunteers. Document everything. And if you’re unsure, ask the FDA for a pre-submission meeting. It’s free, and it can save you millions.
Generic drugs are the backbone of affordable medicine. But they’re not easy to make right. The science is precise. The rules are strict. And the stakes - patient safety - couldn’t be higher.

About Author




Okay but like… have you SEEN the price of levothyroxine? $800 for a 30-day supply? And then some generic comes out at $4 and people are like ‘OMG it’s the same!’ - but then their TSH goes haywire and they’re back in the ER. 🤡
It’s wild how the FDA’s 80/125 rule is basically the pharmaceutical version of ‘close enough for government work.’ I get that it’s statistically sound, but imagine if your car’s engine was allowed to produce 80-125% of the torque it was designed for. You’d be on the side of the road with smoke coming out. And yet, we’re fine with this for drugs that control heart rhythms or seizures. The fact that this system has held up for 30+ years is less a testament to its brilliance and more a testament to how little most people actually monitor their blood levels. We trust because we have to. Not because it’s perfect.
EVERYTHING IS A CONTROLLED EXPERIMENT. The FDA? Controlled by Big Pharma. The ‘healthy volunteers’? Paid addicts who sign NDAs and disappear. The ‘validated assays’? Lab techs who get paid by the company submitting the application. You think they’d let a generic pass if it was actually different? HA. They just tweak the numbers until the software says ‘PASS.’ And the ‘biowaivers’? That’s where the real magic happens - no humans, no data, just a PowerPoint and a prayer. Wake up. This isn’t science. It’s corporate theater.
Biowaivers for eye drops? Fine. But don’t let me catch you swapping generic warfarin for the brand. 😑
Bro, this is why India makes 40% of the world’s generics and still has the FDA’s stamp of approval. We don’t cut corners - we just do it cheaper. 😎 The 80/125 rule? That’s just a way for Americans to overpay. We’ve got labs in Hyderabad running these studies with better precision than some U.S. contract labs. And yes, we use emojis - because science is cool. 🚀🧪
Interesting. I mean, I knew generics were cheaper, but I didn’t realize how much of a minefield the approval process is. Like… you’d think if the active ingredient is the same, it’d be fine. But apparently, the fillers and coating matter more than I thought. Who knew? 🤷♀️
My cousin works at a contract research org that runs bioequivalence studies. She says the volunteers are basically just chill people who need cash and don’t mind being poked every hour for two weeks. They get free meals, a free iPad, and $2,000. Some of them do 3-4 studies a year. It’s like a weird side hustle. The data? Mostly solid. But yeah, one guy once got drunk the night before and they had to scrap the whole run. 😅
USA thinks it owns the world’s drug standards? Hah. We’ve been making safe, effective generics since the 1980s. The 80/125 rule? We follow it better than you. And our labs? More efficient, cheaper, and way less bureaucratic. The FDA approves our drugs because they have to - not because they like us. 🇮🇳💪
Let me get this straight - the FDA lets a generic drug be 25% weaker or 25% stronger and calls it ‘therapeutically equivalent’? And then we wonder why people think the system’s rigged? This isn’t science - it’s a math trick designed to keep drug prices low while pretending safety isn’t being compromised. If your blood pressure med is 15% less effective, you’re not ‘equivalent’ - you’re just one stroke away from a funeral. 🇺🇸💀